Was Sie über Sichelzellkrankheiten wissen müssen
Der Begriff Sichelzellkrankheit (SCD) umfasst mehrere vererbte Anomalien der roten Blutkörperchen. SCD-Patienten haben anormales Hämoglobin, das als Hämoglobin S bezeichnet wird, oder Sichel-Hämoglobin in ihren roten Blutkörperchen.
Hämoglobin ist ein Protein in roten Blutkörperchen, das Sauerstoff im ganzen Körper transportiert.
Der Begriff "Vererbung" zeigt an, dass diese Krankheit durch Gene verursacht wird, die von Eltern an ihre Kinder weitergegeben werden. Im Gegensatz zu Erkältungen oder Infektionen ist SCD nicht ansteckend, so dass gesunde Menschen von Betroffenen nicht der DEZA ausgesetzt werden.
SCD-Patienten erben von Vater und Mutter zwei abnormale Hämoglobingene. Unabhängig von der Art der SCD leidet mindestens eines der beiden abnormen Gene, dass der Körper Hämoglobin S produziert. Inzwischen leidet jemand, der zwei Hämoglobin-S-Gene (oder SS-Hämoglobin) besitzt, an einer Krankheit, die Sichelzellenanämie genannt wird. Sichelzellenanämie ist die schwerwiegendste und häufigste Form der SCD.
SC-Hämoglobin und Hämoglobin-Sβ-Thalassämie sind zwei weitere häufige Formen der SCD.
Einige Formen der Sichelzellkrankheit:
- Hämoglobin SS
- Hämoglobin SC
- Sß0-Hämoglobin-Thalassämie
- Hämoglobin Sβ + Thalassämie
- Hämoglobin SD
- Hämoglobin SE
Wie kann sich eine Sichelzellkrankheit entwickeln??
Zellgewebe des Körpers benötigen Sauerstoff, um richtig zu funktionieren. Normalerweise nimmt Hämoglobin in roten Blutkörperchen Sauerstoff aus der Lunge und transportiert es in alle Körpergewebe.
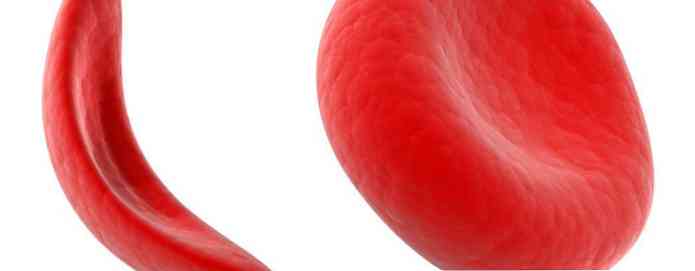
Rote Blutkörperchen, die normales Hämoglobin enthalten, sind wie Stücke geformt (wie Donuts ohne Löcher). Diese Form ermöglicht es den Zellen, flexibel zu werden, sodass sie sich durch große und kleine Blutgefäße bewegen können, um Sauerstoff zu transportieren.
Sichelhämoglobin ist nicht wie normales Hämoglobin. Sichelhämoglobin bildet einen festen Stamm in roten Blutkörperchen, verwandelt sich in eine Sichel oder ist Sichel.
Sichelförmige Zellen sind unflexibel und können an der Gefäßwand hängen bleiben, was Blockierungen verursacht, die den Blutfluss verlangsamen oder aufhalten. Wenn dieser Zustand eintritt, kann Sauerstoff das umgebende Gewebe nicht erreichen.
Ein Mangel an Gewebesauerstoff kann zu starken Schmerzen führen, die als Schmerzkrisen bezeichnet werden. Diese Schmerzen können plötzlich und ohne Vorwarnung auftreten, sodass die Patienten im Krankenhaus effektiv behandelt werden müssen.
Bei den meisten Kindern mit SCD treten keine Schmerzen oder Schmerzkrisen auf, während Jugendliche und Erwachsene an chronischen, anhaltenden Schmerzen leiden können.
Abnormale rote Blutkörperchen und eine zu geringe Sauerstoffzufuhr können auch Organe schädigen, wie Milz, Gehirn, Augen, Lunge, Leber, Herz, Nieren, Penis, Gelenke, Haut oder sogar Knochen.
Da die Form nicht flexibel ist, können Sichelzellen explodieren oder als Hämolyse bezeichnet werden. Der normale Lebenszyklus der roten Blutkörperchen reicht von 90 bis 120 Tagen, während Sichelzellen nur 10 bis 20 Tage anhalten können.
Der Körper bildet immer neue rote Blutkörperchen, um alte Zellen zu ersetzen. Der Körper von SCD-Patienten hat jedoch Schwierigkeiten, die Produktion von neuen Blutzellen mit der schnellen Zerstörung von Blutzellen auszugleichen. Folglich ist die Anzahl der roten Blutkörperchen tendenziell niedriger als die normale Anzahl. Dieser Zustand wird Anämie genannt, als Ursache für die reduzierte Energie des Körpers.
Ob Sichelzellenanämie geheilt werden kann?
Die Sichelzellenanämie ist eine lebenslange Erkrankung. Der Schweregrad der Erkrankung jedes Patienten muss variieren.
In Ländern mit hohem Einkommen wie den Vereinigten Staaten beträgt die Lebenserwartung von SCD-Patienten derzeit etwa 40-60 Jahre. 1973 betrug das Durchschnittsalter der SCD-Patienten in den Vereinigten Staaten nur 14 Jahre. Dies zeigt den Fortschritt bei der Diagnose und Behandlung von SCD.
Derzeit ist die hämatopoetische Stammzelltransplantation (HSCT) die einzige Heilung für SCD. Leider sind die meisten SCD-Patienten zu alt, um sich einer Transplantation zu unterziehen, oder sie haben keine Angehörigen, die eine genetische Übereinstimmung als Spender haben. Geeignete Spender werden benötigt, um die besten Transplantationsergebnisse zu erzielen.
Es gibt viele wirksame Behandlungen, die die Symptome reduzieren und das Leben der DEZA-Patienten verlängern können. Eine frühzeitige Diagnose und regelmäßige medizinische Versorgung zur Vermeidung von Komplikationen tragen ebenfalls zur Verbesserung der Lebensqualität der Patienten bei.